返回主站首页
RSS订阅
|
高级搜索
友好提示:
重庆市艾滋病防治工作宣传教育网开通公众号啦!!!大家快快来围观,及时了解艾滋病最新消息!
艾滋病检测试纸
青年学生群体防艾 同伴教育更有效
艾滋病试纸怎么用
防艾须知——避孕套使用方法
最新研究动态
治疗进展
预防进展
诊断进展
其他
治疗进展
当前位置:
首页
>
大众科普中心
>
科学研究
>
最新研究动态
>
治疗进展
>
艾滋病病毒基因治疗最新进展
上传时间:2024-10-04 21:49:06
文章来源:生物制品公众号
摘要:
早期的基因治疗研究对于遗传性疾病的治愈抱有巨大希望,但各种基因毒性事件的发生导致临床试验暂停,对进展采取了更加谨慎的态度。遗传工程技术的最新进展重新点燃了人们的兴趣,导致2017年首个针对遗传突变的基因治疗产品获得批准。基因治疗(GT)可以在体内或体外进行。体外基因治疗方法具有优势,因为它允许在患者使用前对基因修饰细胞进行表征,并选择所需的特性。在此过程中还可以使用自体细胞,这消除了免疫排斥的可能性。
本综述重点介绍了体外基因治疗的各个阶段、当前研究进展,这些进展提高了这一过程的效率和安全性,以及对人类免疫缺陷病毒(HIV)基因治疗研究的全面总结,其中大多数采用了体外方法。
1.引言
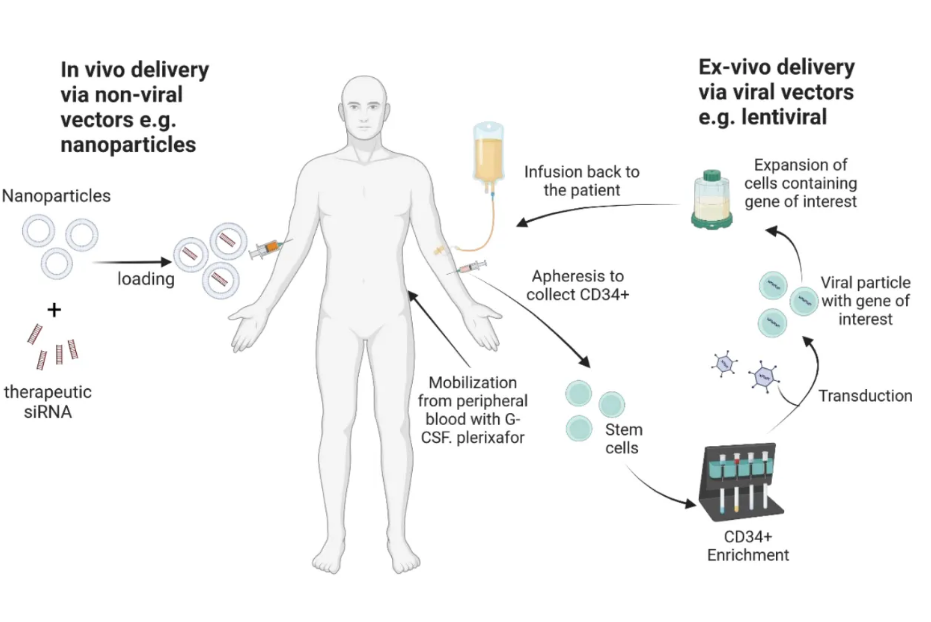
人类基因组的成功测序加深了我们对遗传性疾病和导致慢性及暂时性疾病状况的病理过程的理解。目前有超过5000种基因治疗产品正在针对各种遗传性、慢性和传染性疾病进行试验(https://clinicaltrials.gov/, 访问日期为2023年10月30日)。在开发基因治疗候选药物时,考虑一种有效的递送方法很重要,这将允许感兴趣的基因进行准确处理,在特定作用部位有足够表达,并持续预期的治疗效果,随后降解递送载体而不引起任何不良影响。由于存在引起严重不良反应的潜在危险,如肿瘤形成甚至基因修饰的生殖系传递,开发基因治疗产品(GTP)的过程非常严格,比其他治疗剂需要更长的时间。补充表S1总结了不同监管机构迄今为止批准的基因和细胞产品。最近发表了一篇综述,讨论了其中一些产品的发展。递送感兴趣基因的方法有两种:体内和体外(图1)。体内方法涉及将遗传物质直接递送到患者体内,可以是裸露的基因或封装在颗粒中。另一方面,体外方法涉及从患者或正常供体分离感兴趣的细胞,对它们进行遗传修饰,在某些情况下进行扩增,然后将它们给患者。因此,患者没有直接接触到转移载体。选择,即确保修饰的细胞相对于自然发生的缺陷细胞更优先植入患者体内,可以在给患者施用感兴趣基因之前或之后进行。最近发表了一些综述,强调了基因治疗在各个领域的应用:使用造血干/祖细胞(HSPCs)治疗造血障碍、遗传性皮肤病、神经系统疾病或中枢神经系统遗传性疾病和骨科疾病等。
图1. 体外递送感兴趣基因。使用Biorender.com创建。
2.HIV基因治疗的治愈方法
由于存在对当前治疗有抵抗力的潜伏病毒库,因此获得HIV的治愈一直具有挑战性。这些病毒在转录上是不活跃的,但很大一部分具有复制能力。抗逆转录病毒治疗(ART)可能在潜伏病毒藏身的组织中吸收相对较差,如肠相关淋巴组织(GALT)和中枢神经系统(CNS),这可能有助于这些部位病毒的持续存在。在停止ART后的几周到几个月内观察到病毒反弹。治愈的另一个挑战是HIV对免疫系统的影响:对HIV的体液反应差,中和抗体的产生不足,这通常在感染几个月后才会发生。此外,也有重建HIV特异性CD4+ T细胞的相对失败,这反过来又导致CD8+
细胞毒性T细胞
功能的降低。
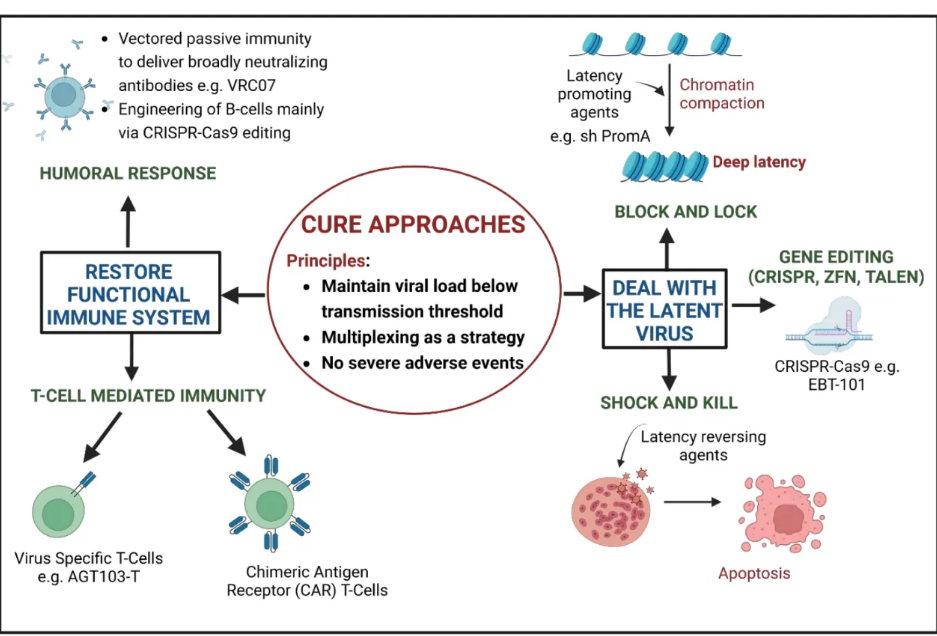
因此,可以合理假设有效的治愈将旨在使潜伏病毒失活或移除,并重建免疫系统。基因修饰的细胞也应该能够抵抗持续的感染,治疗本身不应该引起严重的不良事件。为了重建或增强免疫系统的功能,已经在B细胞和T细胞的遗传工程方面取得了几项进展,包括嵌合抗原受体T(CAR-T)细胞治疗。图2提供了HIV治愈策略的总结。
国际艾滋病学会的多方利益相关者协商就HIV治愈目标产品的最低标准达成一致。临床疗效定义如下:维持病毒载量低于传播阈值(<200 HIV RNA拷贝/mL),在研究的20%或更多人群中有效,平均每年复发率低于10%,缓解期超过2年,并且严重不良事件的可能性极小,如果有的话,即研究人群的不到1%。可能的治愈也可能需要结合不同的方法。
图2. HIV治愈策略。CRISPR—成簇规则间隔短回文重复序列;ZFN—锌指核酸酶;TALEN—转录激活因子样效应子核酸酶。使用Biorender创建。
为了实现对HIV的可能治愈,已经采用了四种策略。
第一种方法是干细胞移植,到目前为止已经治愈了六名患者:柏林患者、伦敦患者、纽约患者、“希望之城患者”、杜塞尔多夫患者和日内瓦患者。
这些患者接受了造血干细胞移植以治疗恶性肿瘤。除了日内瓦患者外,所有患者都接受了与人类白细胞抗原(HLA)匹配的供体的干细胞移植,这些供体对于CCR5等位基因中的32个碱基对缺失(CCR5∆32)是纯合子。由于治疗的侵袭性、成本、发病率和死亡率,以及缺乏具有CCR5∆32的匹配无关供体,这种方法无法扩大到更广泛的人群,因此作为一种普遍适用的方法是不可行的。
第二种方法旨在从细胞库中移除所有活性潜伏病毒,实现无需ART即可检测到的血浆载量,因此提供一种无菌治愈。这种策略被称为“休克和杀死”方法,并已在早期阶段试验中进行了广泛研究。尽管目前这种治愈方法的形式并未采用基因治疗,但它已被包括在本综述中,以涵盖所有当前的HIV治愈方法。“休克和杀死”旨在使用潜伏期逆转剂激活潜伏病毒库中的病毒,这些逆转剂激活病毒转录,通过使用并行的ART阻止新细胞的感染,然后通过诱导凋亡、CD8+介导的溶解或通过体液免疫反应来根除感染的细胞。嵌合抗原受体T细胞(CAR-T)细胞也可以被用来增强杀死感染细胞。
第三种策略旨在通过基因编辑根除整合病毒,使用基于核酸酶的工具或工程重组酶。基于核酸酶的工具包括CRISPR(成簇规则间隔短回文重复序列)、TALEN(转录激活因子样效应子核酸酶)和ZFN(锌指核酸酶)。
也可以使用这些基于核酸酶的工具针对CCR5,使CD4+细胞对感染产生抵抗力,以及使用短干扰或短发夹RNA(siRNA或shRNA)沉默CCR5。基因编辑也可以通过工程重组酶进行,特别是针对HIV特异性长末端重复序列(LTR)的重组酶酶(TRE)重组酶。这是一种工程形式的
Cre
重组酶,针对5′LTR内的34个碱基对区域(称为loxLTR),从而在表达这种酶的感染细胞中去除整合的前病毒DNA。
第四种策略是“阻断和锁定”方法,旨在实现功能性治愈,即在不根除潜伏病毒的情况下保持其处于非活动状态,使血浆病毒载量保持在可检测水平以下。这是通过使用潜伏期促进剂永久沉默或编辑潜伏病毒库来实现的,这些潜伏期促进剂通过病毒启动子的抑制性表观遗传修饰“阻断”病毒转录并“锁定”病毒处于潜伏状态。
大多数HIV基因治疗研究使用体外方法。本综述将重点介绍体外基因治疗的各个阶段,并提供当前采用体外或体内基因治疗方法的HIV临床试验的总结。
3.体外基因治疗
在体外基因治疗研究中需要考虑的一个重要因素是将被修改以纠正特定疾病状况或遗传疾病的细胞来源。这些细胞可以是自体的,从患者自身获得,或同种异体的,从匹配的供体获得。自体来源是有利的,因为没有移植物抗宿主病的危险;然而,这可能不适合老年患者或严重疾病状况(例如,HIV,因为细胞可能是新感染的来源)。由于其自我更新和分化成几个谱系的能力,同种异体干细胞来源最近引起了更大的兴趣,尽管它们的使用存在局限性,如排斥、移植物抗宿主病和长期免疫缺陷。在临床前基因治疗研究中,可以从出生组织中获得干细胞,例如脐带血和胎盘。也可以从胚胎组织中获得干细胞,尽管这引发了伦理问题,或从诱导多能干细胞中获得。在临床研究中,可以根据患者的疾病状况从个体患者中获得细胞,或从与患者组织免疫兼容的供体中获得,或从其他细胞来源中获得,这些来源的细胞不太可能引发免疫反应,如由于缺乏或低表达HLA类I和II抗原而具有低免疫原性的胎盘细胞。体外基因治疗的过程包括获得这些感兴趣的细胞,对它们进行修改,并将它们给患者。为了确保修改后的细胞的有效和优先植入,使用各种调节和选择方法。
3.1.造血干细胞的来源
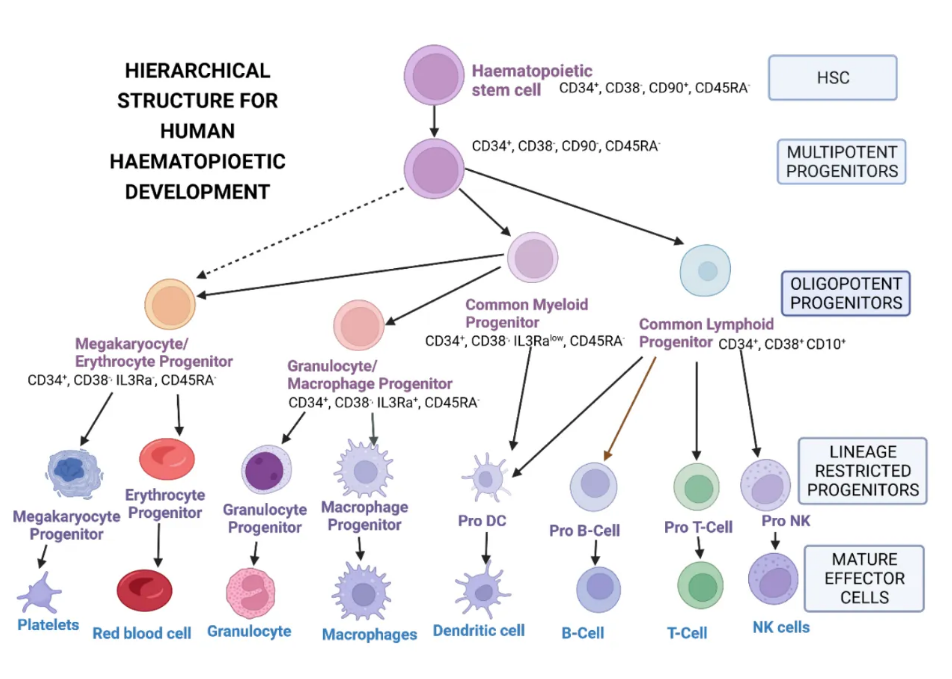
造血干细胞在单基因疾病和免疫系统疾病的基因治疗中很有用。造血干细胞(HSCs)和造血祖细胞(HPCs)负责产生成人血细胞,并以特定标记为特征(图3)。CD34抗原,一种细胞表面粘附跨膜糖蛋白分子,长期以来一直被用作HSCs的标记,以及其他功能,如增强细胞增殖、阻止分化、改善HSCs和HPCs的迁移以及促进淋巴细胞在淋巴组织中与血管内皮的粘附。这些细胞能够分化成各种谱系的功能性血细胞,并且具有自我更新的能力;即,它们可以在不分化的情况下产生女儿HSCs(图1)。
图3. 造血干细胞发展的层级结构。NK-自然杀伤细胞;DC-树突细胞。使用BioRen
der.c
om创建。
3.1.1.骨髓来源的造血干细胞
基因治疗应用中常用的造血干细胞(HSCs)来源包括骨髓、脐带血、外周血,以及最近的胎盘。在骨髓中,成骨细胞影响造血的平衡。原始人类成骨细胞对骨髓来源的CD34+ HSCs的存活至关重要,因为成骨细胞恒定地表达粒细胞集落刺激因子(G-CSF)。同样,HSCs调节成骨细胞分泌IL-6、巨噬细胞炎症蛋白-1α和其他因素,以创造有利于造血的环境。基质衍生因子-1(SDF-1)及其受体C-X-C趋化因子受体4(CXCR4)是建立骨髓的重要决定因素。Tie2,一种受体酪氨酸激酶,也被发现在维持成年骨髓中的HSCs方面不可或缺。CD34抗原具有多个磷酸化位点,其中有两处是蛋白激酶C的位点,一处是酪氨酸磷酸化位点。
Panch等人的文章总结了从骨髓、外周血和脐带血中采集HSCs的方法。简而言之,从骨髓中采集HSCs通常在全身麻醉下进行,大约每公斤体重收集20 mL,且不超过1.5 L的骨髓抽吸液,从髂后或髂前缘收集。在进行该程序之前,可能会从患者身上采集血液,以便在抽吸过程中失去的血液可以重新注入。
3.1.2.外周血来源的造血干细胞
从外周血采集造血干细胞的过程需要几天时间。早期动员方案使用了细胞毒性药物,如环磷酰胺、伊达比星、依托泊苷、铂类和表柔比星;然而,这些药物的使用现在仅限于接受移植治疗恶性肿瘤的患者。目前,在移植患者中,粒细胞集落刺激因子(G-CSF)用于将CD
34
+
细胞动员到外周血中,剂量为5-10 mg/kg/天,持续5至7天,目标是通过白细胞去除术采集至少2×
10
6
CD
34
+
细胞/kg体重。在G-CSF给药期间,血液中HSCs的浓度在3天后增加,大约在第5或6天达到峰值,然后在7天后开始下降。G-CSF的剂量由外周白细胞计数决定,外周CD
34
+
计数是HSCs产量的良好预测指标。产量还取决于年龄、性别、基础条件和G-CSF剂量。G-CSF治疗并非没有挑战,患者可能会经历包括骨痛、头痛、疲劳、肌痛在内的副作用,以及在高风险个体中可能出现心肌梗塞和脑缺血。为了增强G-CSF的动员效果,可以使用AMD3100(Plerixafor,一种可逆的CXCR4拮抗剂)。CXCR4是基质衍生因子1(SDF-1)的受体,具有包括使HSC长期维持静止状态在内的多种功能。Plerixafor以240 µg/kg的剂量给药,在白细胞去除术前4至6小时,发现它可以改善CD
34
+
细胞的采集,尤其是与G-CSF联合使用时。其他HSC动员剂目前正在开发中。
3.1.3.脐带血来源的造血干细胞
从脐带采集细胞涉及切断的脐带静脉穿刺,将血液排入含有抗凝剂的无菌袋中。通常,在过程中收集大约80至160 mL的脐带血(UCB)。CD
34
+
细胞的产量范围为2至7×10
6
/mL UCB,可能受到出生顺序(第一胎>第二胎>第三胎等)、出生体重甚至早期夹闭的影响。这种相对较低的产量限制了它们的应用。
3.1.4.胎盘来源的造血干细胞
人类胎盘最近作为干细胞来源而受到欢迎,因为胎盘干细胞抗原性低,没有伦理限制,并且是多能的。人类胎盘是生殖过程中首先发育的器官,有两个组成部分,胎儿部分(羊膜和绒毛膜)和母体部分(蜕膜)。从受精后6-7天出现到足月的胎盘发育已被详细描述。胎盘的作用是为发育中的胎儿提供营养物质并排除废物,具有各种分泌和免疫调节作用。它可以被划分为以下四个区域:羊膜上皮、羊膜间充质、绒毛间充质和绒毛滋养外胚层。胎盘有多种类型的干细胞,可以分化为造血和间充质组织(脂肪生成、软骨生成、成骨、肝脏、胰腺、肌肉、血管生成和神经生成),表达各种细胞标记,包括OCT4、SOX2和c-KIT等,这些标记也存在于胚胎干细胞中。胎盘HSCs缺乏或表达非常低的HLA类I抗原(HLA-A、HLA-B、HLA-C)和没有HLA类II抗原(HLA-DP、HLA-DQ、HLA-DR),这使它们适用于再生医学以及自体和同种异体移植。与骨髓细胞相比,这些细胞的免疫排斥机会较低,对凋亡有抵抗力,具有增强的细胞增殖和伤口愈合特性,并抑制如TGFβ等促纤维化因子。人类胎盘HSCs是胎儿来源(非母体),并且是CD34+和CD45-dim。用AMD3100(Plerixafor)以300 ug/L的浓度灌注人类胎盘,结果在灌注液中采集到的HSCs数量增加了六倍以上,这些细胞具有集落形成特性,缺乏内皮标记。间充质基质细胞是骨髓造血环境的重要组成部分,与HSCs共培养已被发现可以增强HSCs的增殖,特别是原始的CD34+和CD38-。
已使用脐带血和胎盘来源干细胞的混合物进行造血干细胞移植。已开发出各种协议来分离羊膜上皮细胞、间充质基质细胞和造血干细胞。所有协议都必须克服的一个主要障碍是生产大量具有所需细胞群高度纯度的细胞。
3.2.造血干细胞的分离、纯化和富集
为了确保基因校正细胞的优先植入,重要的是要富集具有构建体的细胞群,并对其进行纯化,以避免在培养过程中使用的蛋白质或其他分子的污染。已使用几种方法来富集HSCs。目前最常见的柱分离技术是德国科隆Milteny Biotech的磁激活细胞分选(MACS®)微珠分离技术(https://www.miltenyibiotec.com/ 于2023年6月4日访问)。在这种封闭系统技术中,微珠(超顺磁性氧化铁纳米颗粒)与CD34抗体偶联,用于磁性标记表达CD34+的细胞。将无菌细胞悬浮液装入MACS®(铁磁性)分离柱中,该柱放置在MACS®分离器的磁场中。在正选择技术中,标记的细胞被保留在柱中,而其他细胞则流过。然后可以从柱中洗脱CD34+细胞,一旦它被从由分离器产生的磁场中移除,就可以用于任何下游应用。也有用于分离CD133+细胞群和CD34+和CD38−细胞的微珠。Baldwin及其同事使用免疫磁珠富集CD34+细胞,然后用荧光激活细胞分选(FACS)对其进行分馏,以富集CD34+和CD38−细胞;另一方面,Radtke及其同事使用FACS分离CD34+、CD90+和CD45RA−细胞,发现这组细胞在表型上是HSC基因治疗的最明确目标,因为这些细胞推动了短期和长期的多谱系植入。他们使用GMP(良好生产规范)级流分选协议来分离这些细胞。这大大增加了所需的复杂性、成本和专业知识。在其他体外研究中,Kays及其同事使用MACS对细胞进行排序,这些细胞除了表达CD34+外,还表达CD105,即转化生长因子-β(TGF-β)受体复合体的组成部分,发现这些细胞富集了具有高植入和重聚能力的早期HSCs,即使在慢病毒转导后也是如此。Laje及其同事用信号淋巴细胞激活分子(SLAM)家族受体分子富集了注定要进行慢病毒转导的HSCs,发现这些细胞被有效转导并且与非转导细胞相比是稳定的。
研究表明,培养造血干细胞(HSCs)超过48小时会降低它们长期植入的能力,建议培养时间不应超过36小时,24小时是一个安全的时间段。Chen及其同事的一项研究发现,使用细胞因子(SCF、TPO和Flt3L)进行体外培养会导致线粒体氧化应激,从而导致丧失干细胞特性,并且CD34+/CD133+中的粘附GPCR G1阳性(ADGRG1+)群体可以在体外培养期间的氧化应激下富集功能性HSCs。
还努力减少培养过程中可能的污染源。重组人血清白蛋白(HSA),是从酵母中产生并用作GMP级组织培养基的组分,可能是其他不直接与白蛋白相互作用的蛋白质甚至色素和小分子的污染源。Wilkinson及其同事最近使用体外和体内小鼠模型发现,聚乙烯醇(PVA)可以作为HSA在培养小鼠和人类HSCs中的替代品,因为它支持HSCs的存活和生长,同时保持表型HSCs特征。PVA培养基中分泌因子的浓度较低,衰老相关基因表达较少,尽管PVA用于人类基因治疗的效果还有待评估。
3.3.使用感兴趣基因修饰造血干细胞
在考虑适当的载体进行基因传递时,需要检查要转导的细胞的特性和载体的特性;是否需要短暂或稳定表达基因修饰;以及在传递到组织时可能导致的“脱靶”效应。这些脱靶效应可能包括宿主-免疫反应和载体本身引起的免疫反应。用感兴趣基因修饰目标细胞可能面临各种挑战。例如,腺相关病毒载体由于阻碍核输入、解壳和第二链合成,因此不能有效转导HSCs。用VSV-G包膜伪装的慢病毒载体具有广泛的细胞嗜性,尽管由于缺乏或表达不足的LDLR受体(它通过该受体进入细胞),原始人类静息T细胞和CD34+细胞不能有效转导。另一方面,T细胞的激活提高了转导效率。在体内使用病毒载体进行基因治疗的情况下,对病毒的预先存在的免疫力可能导致基因修饰细胞数量的减少,这反过来又会限制体内重新填充的速率。转导和重新给药基因修饰细胞之间的时间间隔也至关重要,特别是对于可能分化并限制返回目标部位的干细胞。
为了保护它们的基因组结构,真核细胞具有防御机制,导致外来可移动元素或逆转录病毒的沉默,因此存在诱导沉默机制的可能性,这可能由于缺乏表达而使治疗基因失效。
4.通过病毒载体进行HIV基因治疗传递
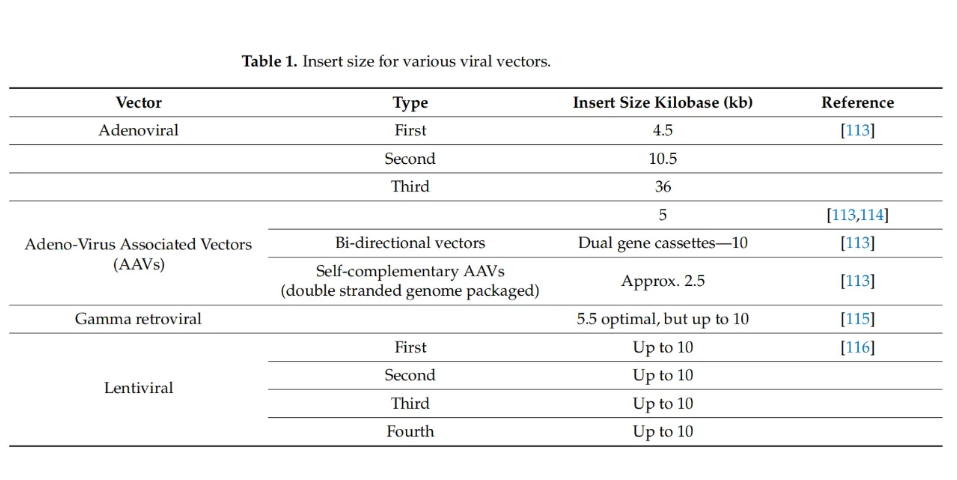
用于体外基因传递的最常见病毒载体是慢病毒载体,因为它能够转导分裂和不分裂的细胞。最初,γ-逆转录病毒载体被用于基因传递;然而,它们靠近原癌基因整合并引起插入性突变的潜力变得明显。从那时起,已经多次尝试使病毒载体更安全、更有效地进行基因传递。选择病毒载体由要转导的细胞类型、病毒载体特性、感兴趣基因(转基因)的大小和期望的效果决定。下表1总结了不同病毒载体的载货量(插入大小)。
4.1.在HIV基因治疗中使用γ-逆转录病毒载体:并发症和提高安全性的方法
γ-逆转录病毒载体是最早用于有效基因治疗的病毒载体之一。这些载体以依赖细胞周期的方式转导活跃分裂的细胞,因此不转导静止的细胞。逆转录病毒最初是首选的载体,因为它们的使用导致转基因稳定地整合到宿主基因组中。然而,这一好处受到大约10%的患者观察到的一系列基因毒性事件的限制,表现为各种形式的白血病。例如小鼠白血病病毒,γ-逆转录病毒更倾向于靠近强增强子区域、转录起始位点、CpG岛和DNase-I高敏位点整合,并可能在靠近原癌基因时引起插入性突变。这些安全问题导致这些临床试验的暂停,因此,已经尝试提高它们的安全档案。现在已知,在逆转录病毒生命周期中,前整合复合体(PIC)与各种宿主转录因子相互作用,特别是溴结构域和外部末端家族蛋白(BET蛋白)和整合酶,导致整合到宿主基因组中。BET蛋白通过将病毒PIC系在宿主染色质上发挥作用,因此,开发不依赖BET的载体可以改变它们的整合档案。事实上,几个小组已经在细胞系和小鼠模型中证明了这种方法的有效性。另一种方法可能是修改整合酶,使其远离靠近原癌基因的整合。与慢病毒载体相比,创建自失活的RV载体的尝试不太成功,因为它们仍然保留着激活癌基因的能力。
4.2.慢病毒载体用于稳定基因表达:挑战和减轻这些挑战的策略
慢病毒是一类逆转录病毒,除了结构基因env、pol和gag外,还有辅助基因,如vpr、vpu、nef和vif,以及调节基因,如tat和rev。最著名的慢病毒是人类免疫缺陷病毒(HIV)。
工程化的慢病毒被认为是适合基因转移的,因为它们可以稳定地转导分裂和不分裂的细胞。与其他逆转录病毒的不同之处在于,大多数逆转录病毒的PIC需要核膜的破裂(在有丝分裂期间),以便允许进入细胞核,而慢病毒PIC通过核孔蛋白和importins的主动运输进入细胞核。PIC是病毒cDNA的组装,一些来自逆转录复合体的病毒蛋白和宿主细胞蛋白。逆转录后病毒DNA的整合是由整合酶和PIC介导的。一项研究发现,PIC更倾向于靶向靠近核孔的开放染色质区域,排除核内部区域和核纤层的周边区域。核周边的转录活跃基因与核孔复合体(NPC)相关,这影响了HIV-1基因表达。慢病毒更倾向于与积极转录的基因整合,整合模式得到目标细胞转录程序的支持。宿主细胞蛋白,晶状体上皮衍生生长因子(LEDGF/p75)直接与病毒整合酶相互作用,没有它,整合酶无法进入细胞核。现在已知LEDGF/p75将整合酶连接到染色质上。
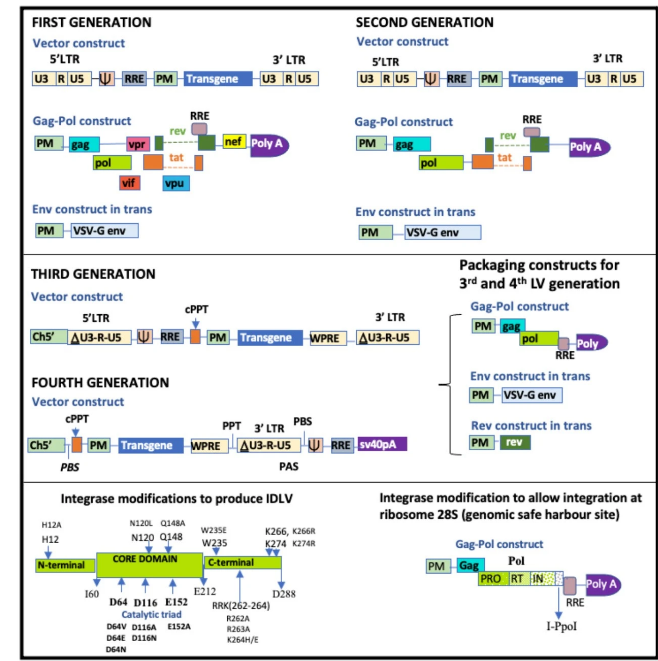
4.2.1.慢病毒载体的演变
自从作为基因治疗载体被发现以来,慢病毒载体(LVs)经历了几次修改(见图4)。第一代LVs包括转基因构建、包膜构建和一个包含gag、pol和所有调节和辅助基因的包装构建。从HIV env改为VSV-G env或任何其他合适的病毒包膜(称为伪型化),允许有效转导多种细胞,尽管VSV-G导致静息淋巴细胞和HSCs的转导效率低下。第二代LVs包括转基因构建、表达VSV-G的包膜构建,以及包含gag、pol和调节基因tat和rev的包装构建。移除了所有辅助基因。第三代LVs在转基因构建中的病毒启动子中进行了修改,其中U3区域通过删除-418至-18的序列部分进行了修改,仅留下18 bp(移除了400 bp),因此创建了自失活(SIN)载体。因此,5′LTR具有18 bp U3、R和U5区域和一个PolyA尾。这使得载体仍然能够携带转基因盒,同时保持转录不活跃。U3被删除的部分被异源启动子替换,通常是巨细胞病毒(CMV)启动子或细胞启动子,如延伸因子1α(EF1α)。3′ LTR、U3区域也被删除,以防止在转染293T细胞过程中通过同源重组与5′LTR重组成完整病毒。载体构建还包含非编码域cPPT(中央多嘌呤区),这提高了RNA被包装进衣壳的效率,以及WPRE(Woodchuck肝炎病毒转录后调控元件),这增强了转基因的转录后处理。包装构建被分割成gag-pol构建和rev构建,删除了tat,因为它的转录激活环已从5′LTR中移除。因此,第三代系统由四个质粒构建组成,如图4所示。三个单独的包装构建降低了在质粒扩增和病毒载体生产过程中形成复制能力慢病毒的重组机会,并减少了启动子干扰相关问题的机会。第三代慢病毒构建通过修改携带转基因的质粒进一步重新设计,可以被认为是第四代慢病毒载体。在这个系统中,称为LTR1或PBS1(引物结合位点1),5′LTR已被移除,RNA信号(PBS-Y-RRE)被放置在3′LTR的下游。因此,这些信号在载体生产过程中存在,但在逆转录过程中丢失,并且不与转导的转基因一起复制,从而进一步提高了安全性。
图4. 慢病毒载体生成的进步。第一代构建包括转基因构建、伪型包膜(通常是VSV-G)和包含gag、pol和所有调节及辅助基因的包装构建。第二代载体与第一代载体类似,只是去除了辅助基因。第三代载体是自失活慢病毒载体,在U3区域有400 bp的缺失。第四代慢病毒载体去除了5' LTR,并将RNA信号(PBS-Y-RRE)放置在3' LTR的下游。RRE:Rev反应元件;PM:细胞来源或其他启动子;cPPT:中央多嘌呤区;VSV-G:泡状口炎病毒G型;WPRE:Woodchuck肝炎病毒转录后调控元件;Y:包装信号。Ch5':嵌合5' LTR;PAS:引物激活信号。
4.2.2.优化慢病毒转导
有效的慢病毒转导人类细胞可能受到人类细胞产生的限制因子的限制,这些限制因子限制了慢病毒感染,如三部分基序蛋白5α(TRIM5α)、tetherin和载脂蛋白B信使RNA编辑酶催化多肽样3(APOBEC3)。其中一个被证明导致慢病毒基因传递到静息记忆T细胞效率低下的因素是限制因子SAMHD1(含α motif和组氨酸-天冬氨酸(HD)结构域蛋白1)。SAMHD1是一种细胞脱氧核苷酸三磷酸水解酶,阻止逆转录。HIV-2编码的vpx基因被发现可以抵消SAMHD1的限制能力。事实上,一项研究发现vpx在他们测试的所有条件下都增加了基因治疗传递,但当基因治疗针对逆转录前的步骤时效果最大。
优化体外培养条件是提高基因转移速率的有用方法。使用Polybrene、鱼精蛋白硫酸盐、Retronectin、热带增敏受体增强子、如Lenti-X accelerator的磁珠、Vectofusin-1、LentiBOOST和Staurosporine等试剂已被发现可以增强转导过程。使用胎牛血清(FBS)在用慢病毒载体转导CD34细胞时并不令人满意,因为它导致祖细胞的转导增加,但HSCs的转导减少。此外,由于对其异种成分产生抗体,存在免疫风险的担忧。已经开发了各种无血清培养基,适用于不同类型干细胞的转导,例如,加拿大温哥华干细胞技术公司的StemSpan无血清扩增培养基(SFEM)。将敲除血清替代品(KSR)培养基添加到StemSpan培养基中,已被发现可以提高原代CD34+细胞和外周血单个核细胞(PBMCs)的转导效率。
4.2.3.慢病毒载体沉默
像γ-逆转录病毒载体一样,尽管程度较轻,慢病毒载体也容易受到转基因沉默和转基因效率变异的影响,这是由于转基因与宿主基因组(染色体位置效应)中其直接基因组邻域的相互作用。一些宿主和载体特征被假设可以引起这种效应,包括DNA甲基化和组蛋白修饰,包括由多梳家族蛋白介导的修饰。可以隔离转基因免受沉默的策略包括使用更强的增强子启动子、脚手架/基质附着区域(S/MARS),这些可以保护增强子免受DNA甲基化,和染色质域隔离子,这些可以抑制抑制性位置效应。
4.2.4.减少插入性突变的策略
已经尝试了各种方法来减少插入性突变的机会。一种策略是将整合导向远离原癌基因的“安全基因组港湾位点”。Schenkwein等人使用I-PpoI,一种来自黏菌Physarum polycephalum的二聚体18-20 KDa同源性内切核酸酶,将整合导向一个高度保守的位点,28S核糖体RNA。这是通过识别该区域的15 bp位点来实现的。另一种策略是通过改变整合酶核心结构域(D64、D116和E152)的催化三联体来生产非整合慢病毒载体。虽然消除了插入性突变的风险,但这些载体只能用于瞬时基因表达。基因表达也低于整合慢病毒载体。修改LEDGF/p75的功能,其功能是指导慢病毒整合,可以允许整合到更安全的区域或更随机的整合,从而减少靠近原癌基因的整合机会。
4.3.腺相关病毒载体(AAVs):限制其使用的挑战
重组腺相关病毒是在临床应用中首选的载体,当需要短暂表达转基因(根据感染细胞的周转时间,从几个月到几年不等)时。这些载体不含病毒DNA,而是基于蛋白质的纳米颗粒,被设计为穿越细胞膜并将DNA送入细胞核。这些载体非常稳定,能够承受操作过程中出现的物理或化学挑战。病毒基因组也容易操纵,并且具有良好的安全档案。AAVs可以携带高达4.5 kb的转基因盒。然而,AAVs的主要挑战是它们在自然界中广泛存在,因此抗AAV免疫很高,12种已知血清型的中和抗体的流行率在某些人群中从20%到100%不等。这些载体的给药也可能引发治疗后的体液反应。正在应用各种策略来克服衣壳免疫、补体固定和AAV基因组感知。一种策略是通过合理设计、随机突变和衣壳重组来工程化衣壳,以防止衣壳中和。另一种策略是免疫抑制,即阻断涉及B细胞激活的经典途径,从而抑制体液反应,尽管体外基因治疗的体液反应可能有限。为了减少AAV基因组感知,可以通过耗尽载体基因组中的CpG二核苷酸来阻止TLR信号传导。腺相关载体可以用来转导多种细胞,包括间充质基质细胞、神经细胞等,尽管它们不适合造血细胞。
许多研究小组正在研究使用AAVs将广泛中和抗体送入骨骼肌,以提供针对HIV的持久效果,无论是作为预防还是治疗,或两者兼有。最近一篇综述文章分析了它们在HIV感染中的使用,以及当前挑战和可以采用的策略来克服其有效性的障碍。
4.4.体外细胞选择和扩增
根据转导的细胞类型、载体类型、感染倍增性(MOI)和使用的转导技术,转导的细胞数量通常低于理论上的预期。因此,转导过程不是100%有效。然而,有几种方法可以最大化转导效率。除了第4.2.2节中提供的例子外,使用高MOI可以产生更高的载体拷贝数(VCN),尽管这增加了异常剪接事件的风险。通过色谱法纯化载体颗粒也可以增加转导效率。转导后,选择携带转基因的细胞并扩增这些细胞以增强它们在体内的植入是有益的。这可以通过细胞表面标记或设计载体表达抗生素抗性基因来实现,然后在应用适当的抗生素时,将允许优先选择携带转基因的细胞。
转导HSC的扩增目标是在保留“干细胞特性”的同时增加数量。通常支持分化为成熟谱系的传统培养基导致自我更新能力的丧失。体外基因治疗的体液反应可能有限。为了限制污染的可能性,最好使用无血清培养基进行HSC扩增。Tajer等人最近的文章总结了添加到培养基中以增强扩增的因素。已经使用了几种细胞因子和生长因子,包括SCF、FLt3配体、TPO(血小板生成素)、IL-3和IL-6,尽管由于IL-3刺激祖细胞的扩增而不是HSCs,它并不包括在某些培养基中。还发现其他化合物也很有用。这些包括前列腺素E2 (PGE2),它通过改善HSCs的归巢和自我更新能力来增强HSCs的植入,和Stem Regenin 1 (SR1),一种芳香烃受体拮抗剂,用于扩增人类和小鼠CD34+,尽管它也被发现更有利于多能祖细胞而不是长期重聚干细胞。这与UM171形成对比,UM171是一种嘧啶哌啶衍生物,它支持干细胞的扩增而不是多能祖细胞。因此,它们可以组合使用,以促进两种谱系的扩增。其他使用小鼠模型的研究表明,可以调节调节干细胞归巢和分化的信号通路。这些包括Notch信号通路、Homeobox基因、Wnt等。锌指转录因子Sall4的过表达导致HSCs(CD34+细胞)在体外和小鼠模型中迅速有效扩增(增加50倍)。
5.基因修饰造血干细胞的给药:增强基因修饰细胞的优先植入
HSC输注后造血重建的第一阶段(最多6个月)是由于承诺的祖细胞(短期HSCs),它们具有有限的自我更新能力。大约6个月后,CD34+克隆减少,第一波克隆重建耗尽,长期HSC逐渐接管重建,它们能够自我更新,其效果甚至在基因治疗后3年仍然存在。因此,短期HSC在早期阶段活跃,而长期HSC在这一阶段进入静止状态,并在短期HSC耗尽时激活。
5.1.清空造血干细胞壁龛的调理方案
在用于造血障碍的自体基因治疗中,目标是用宿主细胞替代有特定遗传缺陷的细胞。在HIV基因治疗中,目标是用有抗性的细胞替代易感染的细胞,或能够对抗病毒的细胞。干细胞壁龛允许维持干细胞并调节其功能;HSC壁龛位于骨髓和脾脏的血管周围。清除用于转导HSCs吸收和消除未转导细胞的壁龛的过程,也称为调理,传统上一直使用化疗和放疗进行,这对目标器官没有特异性。进行调理是为了耗尽壁龛中的造血干和祖细胞,以减少同种异体移植的免疫排斥机会,在恶性疾病的病例中治疗任何恶性肿瘤。调理方案可以分为清髓性调理(MAC)、非清髓性调理(NMA)和减低强度调理(RIC)。清髓性方案由烷化剂组成,有或没有全身照射(TBI),导致全血细胞减少症和1-3周内造血耗尽,不允许自体血液学恢复。因此,为了生存,需要干细胞支持。MAC方案与毒性有关,有时甚至死亡,特别是如果植入出现问题。毒性迹象包括器官衰竭、粘膜炎、骨髓抑制、继发性恶性肿瘤、肌肉骨骼疾病和心脏、肺、生殖和内分泌异常。接受MAC治疗的人群的长期死亡率也高于普通人群。
由于这些不希望的副作用,开发了较低强度的方案。较低的强度意味着可逆的骨髓毒性,与MAC诱导的不可逆骨髓毒性相比。这些方案是NMA和RIC。非清髓性方案引起最小的细胞减少症,不需要干细胞支持。另一方面,减量调理涉及将MAC方案的剂量至少减少30%,虽然它不会导致干细胞的消融,但仍然需要干细胞支持才能在临床上实用。这些调理方案的细节已经回顾过。尽管RIC毒性较小,但发现它对基因治疗的效率较低,因为它导致外周血细胞中基因标记较少,这可能是由于壁龛清除效率低下或对转基因的免疫耐受性诱导不充分。在基因治疗中,辐射或化疗药物的剂量由正在治疗的疾病状况和患者特征决定,包括年龄和性别。欧洲骨髓移植组(EMBT)风险评分是一个用于评估癌症患者移植风险的工具,也可以扩展到任何需要移植的患者。它基于五个因素:患者年龄、疾病阶段、诊断到移植的时间间隔、供体类型(就HLA分型而言)和供体-受体性别组合。通常,30岁以后使用MAC方案死亡率增加,50岁以后不推荐使用。如果供体和受体不是同一性别,也有更高的排斥风险。
5.2.基因修正造血干细胞的体内选择
为了改进携带转基因细胞的选择,在动物模型的体内研究中采用了体内化学选择技术。使用药物抗性基因进行体内选择转基因需要药物抗性基因在转导细胞中高度表达,在非转导HSCs中根本不表达或表达水平低;选择药物应该耗尽大多数非转导细胞,对非造血毒性很小,并且在非恶性条件下不具有遗传毒性。在动物模型中用于选择的基因,特别是在小鼠中,包括多药抗性基因(MDR1)、二氢叶酸还原酶(DHFR)、乙醛脱氢酶(ALDH)、胞嘧啶脱氨酶(
CDD
)、谷胱甘肽S-转移酶(GST)、甲基鸟嘌呤DNA甲基转移酶(MGMT)和次黄嘌呤鸟嘌呤磷酸核糖转移酶(HPRT)。
一项研究使用了O6-甲基鸟嘌呤DNA甲基转移酶(MGMT)的突变体,这是一种DNA烷基化修复酶。突变体MGMTP140K在非人灵长类动物中显示出稳定和有效的HSCs选择,尽管使用(Carmustine-BCNU,例如)的烷基化剂的致癌性质是一个缺点。另一项研究使用短发夹RNA(shRNA)敲除次黄嘌呤鸟嘌呤磷酸核糖转移酶(HPRT),在这些小鼠模型暴露于6-硫鸟嘌呤(6 TG)时实现HSCs的选择。使用这种方法很有吸引力,因为它允许转导的HSCs自我更新、增殖和分化,该方法用于清除壁龛并使携带转基因的细胞优先植入。诱导药物抗性的shRNA仅长48 bp,用于化学选择的6 TG量低,尽管6 TG可能诱发白血性。该组使用MOI为1,在两轮连续病毒暴露后,未用生长因子预刺激以保持“干细胞特性”,实现了20-30%的慢病毒转导效率。
5.3.清除壁龛的最新发展
提高壁龛清除和促进转导细胞植入的最新发展是使用与能够清除壁龛的药物结合的单克隆抗体。这些方法允许针对性地清除HSC壁龛,因此减少了上述调理方案非特异性相关的副作用,并使携带转基因的细胞优先植入。例如,为了改进HSCs的选择,最初的小鼠研究使用了CD45-抗体药物偶联物(CD45-ADCs),但很快意识到这些对HSCs并不特定,并导致淋巴细胞耗尽,因为CD45也存在于淋巴细胞和其他白细胞表面。使用CD117-抗体药物偶联物(CD117-ADC),可以更具体地针对HSCs,在小鼠模型中使内源性HSCs耗尽超过99%。2019年报道这种技术效果的第一组使用链霉亲和素连接子将CD117与毒蛋白连接起来,并测试了其对NOD SCID Gamma(NSG)小鼠和非人灵长类动物的效果。另一组使用了抗人CD117 IgG1单克隆抗体偶联物AMG 191,发现其在非人灵长类动物中有效,目前正在进行1/2期临床试验。另一组将CD117+与鹅膏毒素(MGTA-117)偶联,发现其在患有急性髓细胞性白血病(
AML
)的小鼠模型中非常有效(耗尽了超过95%的HSPCs)。CD117在AML细胞中高度表达,这种方案被发现可以减少这些小鼠的肿瘤负担。
6.基于体外HIV基因治疗的临床试验
如前所述,HIV的基因治疗努力集中在使免疫系统更擅长履行其功能,或旨在沉默或编辑整合的原病毒,使其复制能力不足或使细胞对感染有抵抗力。已经进行了几项临床试验,旨在检查HIV基因和细胞治疗产品的效用和安全性。
6.1.基于细胞治疗的HIV临床试验
最早的细胞治疗研究之一创建了嵌合T细胞受体:CD4-Zeta基因修饰T细胞。这项早期临床研究(NCT01013415)的结果发现,尽管病毒库在各组之间没有差异,但在接受了基因修饰细胞的组中,HIV负担从基线有所下降,并且复发性病毒血症的患者趋势减少。AGT103-T是一种产品,可以恢复慢性HIV病患者的gag特异性CD4+ T细胞反应。最初的1期临床试验结果(NCT03215004)显示了一些有希望的结果。对参加研究的患者进行了白细胞去除术。一旦细胞被基因修饰并通过了质量控制测试,患者在基因治疗产品输注前一周用环磷酰胺进行了非清髓性调理,剂量为1 g/m
2
。AGT103-T修饰细胞以每公斤体重2到2100万细胞的剂量输注。即使在基因治疗产品输注后6个月,也可以观察到细胞的基因标记。没有记录到严重不良事件。
SB-728是一种锌指核酸酶(ZFN)介导的,CCR5修饰的,自体CD4+ T细胞产品。这项研究的各种版本发现,即使在重复剂量给药后,也没有发现严重不良事件。当使用环磷酰胺进行调理时,植入效果更好。其中一项研究发现,尽管病毒反弹有延迟,并且CD8+ T细胞反应改善了6个月以上,但对潜伏病毒库没有长期影响。另一项研究,NCT03617198,将CAR-T细胞治疗与ZFN CCR5修饰相结合,作为一种双重治疗方法,确保CD4+细胞对感染有抵抗力,并且同时能够检测和清除HIV感染的细胞。最后,NCT04648046使用了编码双特异性抗gp120 CAR分子的慢病毒载体(LVgp120duoCAR-T)来针对表达HIV gp-120的细胞。
6.2.基于通过逆转录病毒载体进行基因传递的临床试验
早期直接针对HIV的研究使用了逆转录病毒载体来传递抗HIV基因。在一项研究中,使用了同种基因T细胞(来自同卵双胞胎),使用逆转录病毒载体传递了细菌基因NeoR(NCT00001353)。这项研究的目的是证明,通过基因转导的同种基因细胞实际上可以通过成熟细胞的分裂,而不是前胸腺干细胞的分裂,持续数周至数月。在涉及双胞胎的一项研究中(NCT00001535),从血清阴性的双胞胎中获得了同种基因CD4+淋巴细胞,并用携带反义TAR或反义Tat/Rev RNA,反式优势Rev蛋白或两者结合的逆转录病毒载体转导。临床前研究表明,所有使用的抗HIV载体都抑制了HIV,反式优势Rev蛋白显示出更大的抑制作用。尚未发布临床试验的结果。涉及双胞胎的所有研究的长期随访(Gemini Study—NCT04799483)目前正在进行中。另一项使用逆转录病毒载体的研究,NCT00002221,从计划进行自体骨髓移植的非霍奇金淋巴瘤HIV阳性患者中获得了外周血CD34+细胞,并将这些细胞分成三个池。一个细胞池用含有两个核糖酶序列“L-TR/Tat-neo”的逆转录病毒载体转导,第二个池用对照载体‘LN’转导,第三个池保持未修饰细胞。然后将这些细胞重新输注回患者体内。尚未发布这项研究的结果。除了逆转录病毒载体观察到的插入性突变风险外,这项后续研究的成功也可能因携带治疗基因的细胞比例低而受到阻碍。
RevM10是HIV-1 Rev基因的优势负性突变体。使用逆转录病毒载体将这个基因转导入CD4+ T细胞,结果抑制了HIV复制,并持续了6个月。尚未发布在造血干细胞中研究RevM10的结果(NCT00003942)。逆转录病毒载体也用于传递MazF,一种Tat依赖性内切核酸酶基因,在1期研究中评估其安全性、耐受性和免疫原性。MazF源自大肠杆菌,选择性地切割mRNA中的ACA序列,这些序列在HIV中很常见。这项研究发现,单次静脉注射用MazF转导的自体CD4+ T细胞导致CD4+和CD8+细胞增加,这种效果至少持续了6个月。
核酸酶是催化RNA分子,可以针对任何具有NUX切割位点的RNA序列,其中N是任何核苷酸,X是A、C或U中的任何一个。在一项概念验证的1期研究中,Rz2,一种针对tat基因起始密码子附近GUA序列的核酸酶,通过逆转录病毒载体传递到同种基因外周血单个核细胞(PBMCs)中,这些细胞是从血清阴性双胞胎的基因修饰细胞中获得的,然后输注到血清阳性双胞胎体内。共有4名患者参加了这项研究,该研究表明基因标记细胞的持续存在,无论是短期(24周)还是长期(44个月),在此期间没有观察到严重副作用。在一项相关的2期基因治疗试验中,通过逆转录病毒载体将一种抗HIV核酸酶转导到自体CD34+细胞中,患者接受了tat–vpr–特异性抗HIV核酸酶(OZ1)或安慰剂。没有与OZ1相关的不良事件。尽管在主要终点(大约48周)时OZ1和安慰剂组之间的病毒载量没有统计学上的显著差异,但在40周时观察到了显著差异,并且直到100周,OZ1组的CD4+ T细胞高于安慰剂组。在长期随访研究中,NCT01177059,最初入组的68人中有18人完成了研究,其中7人有严重不良事件,包括各种类型的肿瘤(例如,基底细胞癌、霍奇金病、卡波西肉瘤、乳头状甲状腺癌、皮肤癌),这些最有可能是由逆转录病毒载体引起的。
6.3.基于慢病毒载体的HIV研究
在VRX496(Lexgenleucel-T)研究中,使用携带针对HIV包膜的抗HIV反义基因的基于HIV的慢病毒载体修饰自体T细胞。安全性和耐受性研究,NCT00295477,发现血液中的植入半衰期为5周。一些患者的基因标记细胞甚至在5年后还存在。这项研究表明,基因修饰细胞可以对HIV施加遗传压力。
另一项使用慢病毒载体传递抗HIV基因的研究结合了针对CCR5的短发夹RNA(shRNA),它阻止病毒进入,人/恒河猴嵌合TRIM5α,一种在进入细胞质时破坏病毒衣壳解壳的天然分子,因此抑制HIV感染,以及转录激活反应(TAR)诱饵,它通过结合病毒Tat并将其隔离,因此阻止其介导前病毒DNA的有效转录。这项临床试验的结果尚未发布。
在一组研究中,接受自体移植治疗淋巴瘤的患者被输注了用含有tat-rev shRNA、TAR诱饵和CCR5核糖酶(LV Rhiv7-shITAR-CCR5RZ)的慢病毒构建体修饰的CD34+造血干细胞。四名患者的早期结果表明,基因修饰细胞在输注后第11天植入,并且即使在24个月时,载体仍然存在,并且在病毒血症后选择基因修饰细胞。尚未发布各个研究的结果。
Cal-1是一种双重抗HIV基因转移慢病毒构建体,它传递一种沉默CCR5的shRNA(sh5),并在不同的启动子下,编码一种小肽抑制剂的序列,这种抑制剂可以防止HIV包膜与宿主CD4+细胞的融合(C46)。已在不同地点对Cal-1进行了几项研究。在NCT01734850中,对13名患者进行了Cal-1治疗,并评估了该治疗的安全性。Cal-1的临床前研究表明,该治疗有效且安全。在临床试验中,使用白消安调理来清除壁龛,以允许基因修饰细胞的植入。在两个接受白消安调理的治疗组中观察到严重和危及生命的不良事件,与单剂量白消安相比,双剂量白消安的副作用更严重。未接受调理的组没有严重不良事件。患者目前正在进行长期随访研究(NCT02390297)。与调理相关的毒性是成功传递基因治疗的常见挑战,因此开发更安全、更有效的方法来清除壁龛,以实现成功和稳定的植入是值得的。
这些临床研究表明,在实现足够植入的足够清除壁龛、使基因修饰细胞的生产和由于调理导致的骨髓毒性之间存在微妙的平衡。
为了帮助推进HIV治愈方法的临床前研究,例如使用si/shRNA的“阻断和锁定”方法的研究报告,解决这些挑战至关重要。这组临床前小鼠研究已经报告了通过si/shRNA成功沉默HIV,尤其是shPromA,它针对病毒启动子中的串联NF-KB转录因子位点,以诱导下游病毒基因表达的转录基因沉默和抑制性表观遗传修饰(见图2)。结合含有CCR5 shRNA的Cal-1载体,目前正在研究针对启动子的si/shRNA PromA,以开发针对宿主CCR5和病毒启动子靶标的组合HIV基因治疗。
7.基于体内传递的HIV基因治疗临床试验
采用体内方法治疗HIV的研究主要处于临床前阶段。体内方法涉及直接将基因治疗产品通过病毒载体(如腺病毒载体、腺相关病毒载体或非整合慢病毒载体)或非病毒方法(如纳米粒子)引入患者体内。体内方法可能无法提供持久的治愈,因为没有转基因与宿主遗传物质的稳定整合,并且需要优化特定细胞的传递,以减少脱靶效应的机会。通过重组酶或核酸酶系统的基因编辑,或使用RNA干扰或可能的CRISPR干扰的沉默机制,可以使用体内方法。
目前的临床试验包括使用leronlimab的研究,leronlimab是一种抗CCR5人源化IgG4抗体,通过与CCR5相同的附着位点(细胞外环2和N-末端结构域)结合,竞争性地抑制HIV env附着到CCR5。这种药物通过皮下给药,该组目前正在使用合成AAV载体进行传递。这项研究的几个版本—NCT00642707、NCT02175680、NCT02355184、NCT02483078、NCT02990858、NCT03902522、NCT02859961和NCT05271370—证明了产品的安全性和有效性,患者大多经历了轻微的副作用,并且在较高剂量的525 mg和700 mg时观察到效果。
CRISPR-Cas9技术
用于HIV治愈
CRISPR及其CRISPR相关核酸酶9(Cas9)系统是一种基因编辑系统,通过导向RNA介导的基因组区域的双链断裂,然后通过非同源末端连接途径进行修复。最近发表了几篇描述HIV临床前研究的综述文章[222,223]。最近使用CRISPR-Cas9治疗HIV的临床试验是NCT03164135,该试验使用这项技术在进行异体干细胞移植治疗血液恶性肿瘤的患者中敲除HSCs中的CCR5基因。一名急性淋巴细胞性白血病患者的初步结果显示,完全供体嵌合体的缓解,并且敲除CCR5的供体细胞持续了19个月,没有基因编辑不良事件。然而,携带修改的淋巴细胞的百分比仅为5%,团队正在研究如何使过程更有效。另一种基于CRISPR-Cas9的HIV基因治疗产品是EBT-101(NCT05144386)。这种基因治疗作为单次静脉注射给药,并通过使用两个导向RNA靶向整合原病毒的三个位置的AAV传递。最近有三名患者参加了1期临床试验,该公司(Excision Bio Therapeutics, San Francisco, CA, USA)在2023年10月的欧洲基因和细胞治疗年会上提出的初步结果显示,没有剂量限制毒性或严重不良事件。
CRISPR-Cas 9可以用来靶向HIV-5′LTR,这是启动子区域,因此可以阻止潜伏病毒的复制或激活;CCR5或CXCR4区域,使细胞对感染有抵抗力;或促进HIV复制的限制因子,阻断它们的活性。虽然目前还没有CRISPR-Cas9基因治疗被批准作为HIV治愈方法,但FDA最近批准了第一种用于镰状细胞病的基于CRISPR-Cas9的细胞基因治疗方法,Casgevy(
https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease
, 访问日期为2024年1月27日),突显了基因治疗的创新进步和CRISPR-Cas9作为HIV治愈方法的潜力。
8.结论
长期或永久表达抗HIV基因和修改CD4+和CD34+细胞以使它们对感染有抵抗力或允许破坏HIV生命周期是实现HIV治愈的重要策略。尽管联合抗逆转录病毒治疗的出现大大改善了患者的治疗结果和HIV感染者的生活质量,但治愈是可取的,特别是对于已经对当前方案产生耐药性的患者。至少提供功能性治愈的产品可能需要多重治疗剂与不同作用模式的组合,以最小化耐药性的机会,并且是长效的,或理想情况下需要单次给药。因此,至关重要的是要利用当前生物技术的进步来增强基因治疗方法,并使基因治疗产品更安全、更经济、更持久。
[字体
大
中
小
]